
A well-structured regenerative medicine workflow is the difference between a therapy that reliably reaches patients and one that stalls in development or fails at the bedside. The field is growing fast. The regenerative medicine market is projected to expand from $16 billion in 2023 to $49 billion by 2028, yet clinical translation remains difficult. For healthcare professionals and researchers working in this space, understanding how each stage of the process connects — from cell sourcing through quality release — is what makes a program functional, scalable, and compliant.
Table of Contents
- Key takeaways
- Prerequisites for starting a regenerative medicine workflow
- Step-by-step execution of the workflow
- Troubleshooting and common challenges
- Verification, quality assurance, and regulatory compliance
- What we have learned from years of regenerative workflows
- How Nortex Tissue Regeneration supports your regenerative medicine goals
- FAQ
Key takeaways
| Point | Details |
|---|---|
| Workflow structure determines outcomes | A defined, sequential regenerative therapy process reduces errors and improves reproducibility across patient batches. |
| Material sourcing decisions have long-term consequences | Choosing defined, animal component-free reagents early supports a smoother GMP transition later without costly revalidation. |
| Chain-of-identity is non-negotiable for autologous work | Every autologous batch represents a single patient event. Zero-tolerance protocols for labeling and handling errors are required. |
| Conditional release enables timely delivery | Rapid testing methods such as Gram staining allow product release when sterility test timelines exceed product shelf life. |
| Digital QMS integration supports compliance | Electronic batch records and automated monitoring are no longer optional. They are the backbone of defensible data integrity. |
Prerequisites for starting a regenerative medicine workflow
Before the first cell is collected, the infrastructure around your regenerative medicine workflow needs to be in place. This is where many programs struggle. The excitement around the biology frequently outpaces the operational readiness, and the cost of correcting that gap later is significant.
Material and cell source requirements
The choice between autologous and allogeneic sources shapes everything that follows. Autologous workflows use patient-derived cells, which means each batch is unique, the manufacturing process scales out rather than up, and the tolerance for handling errors is effectively zero. Allogeneic programs draw from a donor population, allowing for more conventional batch manufacturing and earlier standardization.
| Requirement | Autologous | Allogeneic |
|---|---|---|
| Donor eligibility testing | Limited; traceability mandatory under 21 CFR §1271 | Extensive infectious disease testing required |
| Batch size | Single patient event | Multi-patient lots possible |
| Chain-of-identity controls | Strict, zero-tolerance | Less patient-specific; lot-level traceability |
| Scalability model | Scale-out | Scale-up |
| Raw material standardization | Critical from day one | Equally critical; earlier comparability leverage |
Beyond the source decision, your material inputs need careful selection. Using undefined or animal-derived components in early development creates a significant problem later. Changing culture components mid-development forces formal comparability validation to prove equivalence, which costs time and resources that most programs cannot afford. Define your raw materials early and stick with them.
Key infrastructure requirements before initiating a regenerative therapy process:
- Cleanroom or ISO-classified environment appropriate to the therapy type and regulatory classification
- Validated equipment with documented calibration and maintenance schedules
- Chain-of-custody and chain-of-identity tracking systems, ideally electronic
- Qualified reagents and culture media with certificates of analysis and traceability documentation
- SOPs covering donor screening, labeling, processing, storage, and release criteria
Pro Tip: Implement your chain-of-identity tracking system before you run your first batch, not after. Retrofitting traceability onto an existing process is far harder than building it in from the start.
Step-by-step execution of the workflow
A structured stem cell treatment procedure or tissue engineering workflow follows a clear sequence. Deviating from it without documentation is where programs introduce risk. Here is how the process typically progresses in a well-run program:
-
Patient or donor screening and consent. Confirm eligibility criteria, document informed consent, and assign a unique patient or lot identifier that travels with the product throughout the entire process.
-
Cell collection and receipt. Harvest the biological starting material, whether bone marrow aspirate, adipose tissue, peripheral blood, or another source. Log receipt time, condition, and chain-of-custody transfer in your tracking system immediately.
-
Initial processing and isolation. Separate the target cell population using centrifugation, density gradient separation, or magnetic selection, depending on the cell type. Document processing parameters and any deviations.
-
Cell expansion and culture. Expand cells to the required dose using defined, validated culture conditions. Monitor growth kinetics, morphology, and phenotype at defined intervals. This is where your choice of culture format, planar versus bioreactor, has a real impact on yield consistency.
-
Quality control sampling. Pull samples at defined points for sterility, mycoplasma testing, identity confirmation, viability assessment, and potency assays where applicable. The tissue microenvironment the cells will encounter post-administration is itself a variable, so understanding your product’s functional capacity matters here.
-
Formulation and fill. Bring cells to final concentration in the appropriate excipient. For cryopreserved products, add cryoprotectant and fill into labeled final containers that maintain chain-of-identity.
-
Conditional release or hold pending testing. For time-sensitive therapies such as autologous CAR-T where rapid tests like Gram stain enable release while the 14-day sterility test continues post-infusion, conditional release protocols should be pre-approved and clinical management triggers documented for any positive results.
-
Delivery and administration. Transfer the product to the clinical team under defined temperature and time conditions. Confirm receipt and administer according to protocol.
-
Post-administration documentation. Record outcome data, adverse events, and any deviations. Close the batch record and archive according to your document retention policy.
Pro Tip: Treat each batch record as a legal document from the moment the product identifier is assigned. Incomplete or amended records are one of the most common findings during regulatory inspections, and they are entirely preventable.
A personalized medicine approach like autologous therapy demands that every step in this numbered sequence is traceable, timestamped, and independently verified where possible.

Troubleshooting and common challenges
Even well-designed workflows encounter problems. The variability inherent in patient-derived cells is the most difficult challenge in a regenerative therapy process. A 70-year-old patient with osteoarthritis will not yield the same cell quality as a 30-year-old athlete. That biological reality has to be accounted for in your specifications, not ignored.
Common pitfalls that compromise workflow integrity:
- Chain-of-identity errors. Mislabeling at any point is catastrophic in autologous programs. Each batch is a single patient event, and there is no margin for mix-ups. Two-person verification at every labeling step is the standard, not the exception.
- Using undefined reagents in early development. This is the single most avoidable source of GMP transition pain. If your current media contains animal serum, the clock is running on a comparability study.
- Underestimating sterility testing constraints. For short-shelf-life products, a 14-day sterility test result arrives after the patient has already been treated. Conditional release protocols need to be in place before this situation arises, not during it.
- Scaling without standardization. Treating scale-out as simply running more parallel processes without formal process characterization leads to inconsistent yields and potency variability.
- Siloed teams. Manufacturing, QA, and clinical teams that do not communicate in real time create handoff failures. This is where documentation gaps and chain-of-identity breaks tend to occur.
Platform-based modular approaches are widely recommended for scaling regenerative medicine programs because they build reusable process layers rather than designing everything from scratch for each new therapy. Applying that thinking early reduces both development time and regulatory burden.
Cryopreservation deserves specific attention. Freeze-thaw validation should be performed with worst-case conditions representing the actual shipping and storage scenario your product will face, not idealized laboratory conditions.
Verification, quality assurance, and regulatory compliance
Quality assurance in a regenerative medicine workflow is not a final step. It is a continuous function woven through every stage. The GMP proportionality principle for advanced therapy medicinal products allows risk-based flexibility, which is useful for patient-specific small-batch manufacturing. However, that flexibility requires robust documentation to justify every decision made.
| Regulatory Aspect | Autologous Therapies | Allogeneic Therapies |
|---|---|---|
| Regulatory classification | Often 351 HCT/P or IND required | Typically BLA or IND required |
| Donor eligibility testing | Limited; traceability under 21 CFR §1271 mandatory | Extensive; infectious disease markers required |
| GMP requirements | Phase-appropriate; risk-based flexibility allowed | Full GMP expected earlier in development |
| Release strategy | Conditional release acceptable with protocol | Full QC testing before batch release standard |
| Regulatory flexibility | FDA CMC flexibilities for early-phase programs | More prescriptive for large-scale manufacturing |
Batch release documentation needs to capture every critical quality attribute tested, reference the approved specification, and record the identity of who authorized release. Electronic batch records, when properly validated, reduce transcription errors and provide audit trails that paper systems cannot match.
From a regulatory standpoint, understanding whether your product falls under minimal manipulation definitions in 21 CFR 1271.10 or requires full BLA submission changes your compliance obligations substantially. This determination should happen before you finalize your manufacturing approach. Digital QMS platforms with electronic batch records and automated environmental monitoring are now considered standard infrastructure for programs seeking to demonstrate data integrity to both the FDA and EMA.
What we have learned from years of regenerative workflows
I want to say something that most workflow guides skip over. The biggest mistakes I see in regenerative programs are not technical. They are organizational.
The most common scenario is a research team that has developed a genuinely promising therapy, built it around a bespoke process, and then realized at the IND-enabling stage that their culture system includes an undefined serum component and their documentation is essentially lab notebooks. The science is real. The workflow is not transferable.
What I have found is that early decisions about culture format carry consequences that last years. The choice between a planar flask system and a bioreactor is not just a scale decision. It determines your process variability, your monitoring strategy, and your comparability burden if you ever need to change it. Making that choice deliberately, with GMP transition in mind, costs almost nothing. Making it reactively costs months.
I have also noticed that teams often treat QA as an external check rather than an internal voice. The programs that consistently produce clean batch records and pass inspections are the ones where QA is in the room during process development, not just at the end. That integration is a cultural choice as much as an operational one.
The how does regenerative medicine work question gets asked constantly, and the honest answer is that it depends on a complex interplay between cell quality, delivery, and the patient’s tissue environment. Building a workflow that respects that complexity, rather than oversimplifying it, is what separates programs that generate meaningful clinical data from those that generate noise.
— Felix
How Nortex Tissue Regeneration supports your regenerative medicine goals
At Nortextissueregeneration, we apply the same principles of structured, evidence-based process design to every patient we work with. Whether you are exploring stem cell therapy for chronic joint degeneration or considering PRP therapy for sports injury recovery, our approach is grounded in documented protocols, personalized assessment, and realistic expectations about outcomes. We work without surgery, without long recovery periods, and without assumptions. If you are a clinician or researcher looking to understand how structured regenerative therapy translates from a manufacturing context into real patient care, we welcome the conversation. Reach out to our team to learn how we can support your clinical or research objectives.
FAQ
What are the main stages of a regenerative medicine workflow?
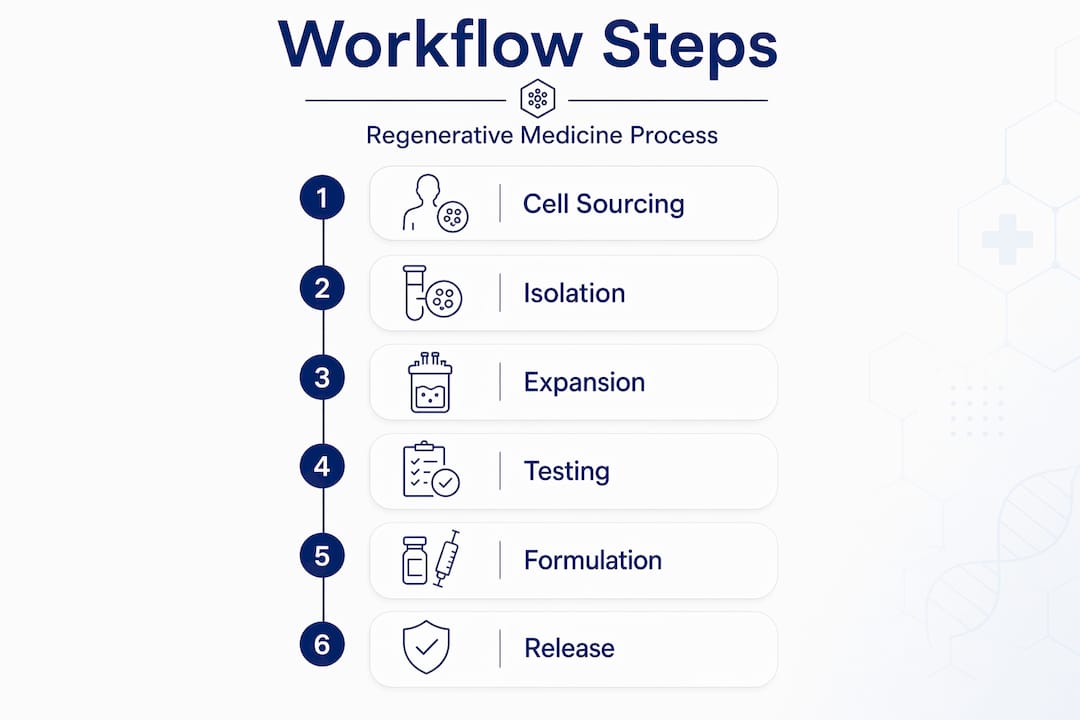
A regenerative medicine workflow moves through cell sourcing, isolation, expansion, quality control testing, formulation, and final delivery to the patient. Each stage requires documented protocols and traceability systems to protect product integrity.
Why does chain-of-identity matter in autologous stem cell procedures?
In autologous therapy, each batch corresponds to a single patient. Any labeling or handling error cannot be corrected by pulling from another lot, so two-person verification at every step is the standard practice.
What is conditional release in a stem cell treatment procedure?
Conditional release allows a product to be administered before all quality control tests, such as the 14-day sterility test, are finalized. Rapid tests like Gram staining confirm safety at release, and clinical management protocols are triggered if post-infusion results are positive.
How do FDA regulations affect regenerative therapy process design?
The FDA uses a two-tier system based on minimal manipulation and homologous use criteria. Products that do not meet those criteria require IND or BLA submissions, and CMC flexibilities allow phase-appropriate GMP in early clinical stages.
When should GMP-compliant materials be introduced into a tissue engineering workflow?
GMP-compliant, animal component-free materials should be introduced as early as possible in development. Replacing undefined reagents later forces comparability studies that can delay regulatory submissions and add substantial cost.
Recommended
- The Complete Regenerative Therapy Checklist for Joint Pain
- PRP vs Stem Cell Therapy: Which Is Right for You? – Nortex Tissue Regeneration
- Outpatient Regenerative Therapy: Evidence, Options, and Real Outcomes
- How Is PRP Therapy Made? From Blood Draw to Injection Step-by-Step – Nortex Tissue Regeneration



